ISSN : 2471-8041
Medical Case Reports
Janet Bolaji and Tim Wang
International Conference on Physicians, Surgeons and Case Reports
November 19-20, 2018 Paris, France
Frimley Park Hospital, UK
Frimley Park Hospital, UK
ScientificTracks Abstracts: Med Case Rep
DOI: 10.21767/2471-8041-C2-005
Abstract
Introduction: A 46 year old female was referred to the metabolic medicine clinic for evaluation. She was referred via her general practitioner with persistently low alkaline phosphatase (ALP) levels which were first noticed during an occupational medical assessment. There was no personal history of fractures or arthritis and no family history of fractures or dental issues. Commonly considered conditions associated with low serum ALP include coeliac disease, micronutrient deficiency, Wilson’s disease, drug therapy, anaemia and hypothyroidism.
Results: On review, serum ALP was noted to be chronically between 15-20 IU/L (reference range 30-130 IU/L) and this had been the case for as far back as results were available which was preceding 13 years. Laboratory testing for free T4 and TSH were normal. In addition, tests for serum copper, ceruloplasmin levels, tissue transglutaminase antibodies, vitamin B12, folate and magnesium levels were all also unremarkable. Zinc was had been previously low but she had been started on supplementation and levels had normalised by the time of review. Serum vitamin D was mildly low at 59 nmol/L (reference range is 75-200 nmol/L) and supplementation did not affect her low serum ALP levels. X-ray imaging of the knees revealed no evidence of chondrocalcinosis. Serum pyridoxal phosphate levels were elevated at 161.4 nmol/L (reference range 35-110 nmol/L) and also combined with low alkaline phosphatase which was suggestive of primary hereditary hypophosphatasia. This patient was referred to a tertiary centre, and urinary phosphoethanolamine was deemed unnecessary, since it is often normal in patients with hypophosphatasia. This patient went on to be referred for genetic testing and family screening.
Conclusion: Primary hereditary hypophosphatasia was initially considered to be an unlikely diagnosis for this patient since she had been asymptomatic and did not present with the typical bony symptoms or complications. Monitoring is usually recommended in patients with primary hereditary hypophosphatasia and thus, recognition of the condition can have important implications for patients and their families. Therefore, although uncommon, it is a key diagnosis to bear in mind.
Biography
Janet Bolaji has completed her BSc in Physiology at King’s College London in addition to her primary medical qualification from Swansea University. She completed her Foundation Training at Frimley Park Hospital and St Helier Hospital. Her rotations included Chemical Pathology and General Medicine. She has an interest in Endocrinology and Nutritional Medicine and is furthering this interest by completing a Masters’ degree at University College London (UCL). She also has an interest in medical education and enjoys organising teaching sessions for medical students.
E-mail: janetbolaji@yahoo.com
Google Scholar citation report
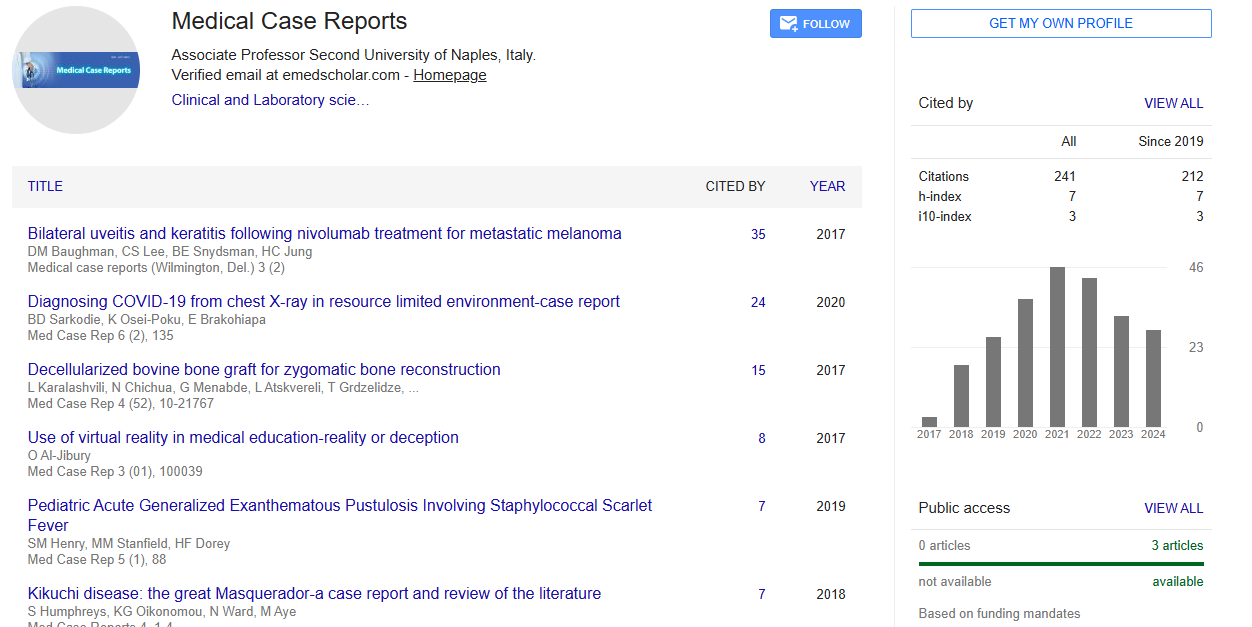
Citations : 241
Medical Case Reports received 241 citations as per Google Scholar report
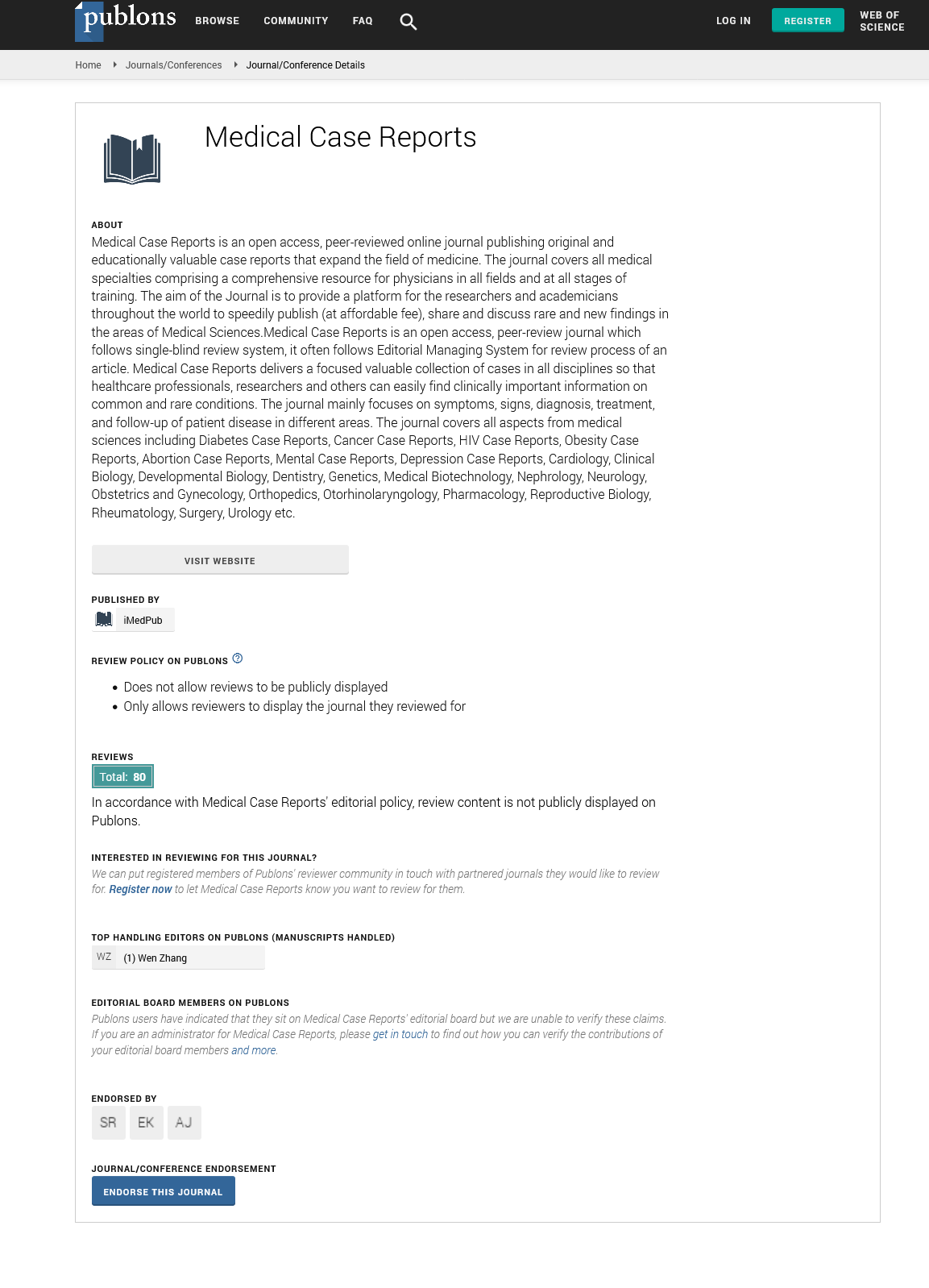
Medical Case Reports peer review process verified at publons
Abstracted/Indexed in
- Google Scholar
- China National Knowledge Infrastructure (CNKI)
- Cosmos IF
- Directory of Research Journal Indexing (DRJI)
- WorldCat
- Publons
- Secret Search Engine Labs
- Euro Pub
Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences