ISSN : 2471-8041
Medical Case Reports
A case report on rare variant of ILNEB syndrome
International Conference on Physicians, Surgeons and Case Reports
November 19-20, 2018 Paris, France
Sumitha Tarur, T Arasar seeralar and S Srinivasan
Institute of Child Health and Hospital-Madras Medical College, India
Posters & Accepted Abstracts: Med Case Rep
DOI: 10.21767/2471-8041-C2-006
Abstract
Congenital interstitial lung disease with nephrotic syndrome and epidermolysis bullosa (ILNEB) is thought to be caused by mutation in the alpha 3 integrin gene (ITGA3). Alpha 3 integrin heterodimerises with beta 1 integrin and functions as a receptor for extracellular matrix proteins. It is important for cellular adhesion and is a component of fetal and adult tissues. To date, only six patients with ILNEB syndrome have been reported. All the patients carried homozygous ITGA3 mutations and presented with severe phenotypes that culminated in death before two years of life. In this report, we present an atypical case of the ILNEB syndrome. A nine-year-old female child, second born of third degree consanguineous parents, came with complaints of insidious onset breathlessness for the past six months. Antenatal, natal and perinatal period were uneventful. The child was apparently normal till two years of age after which she developed blistering skin lesions that healed with scarring. She also had a history of passing foamy, frothy urine since the fourth year of life. On examination she had normal mentation, short stature, toe nail dystrophy, scarring at rest alopecia and epihora. She was tachypenic but could do her activities of daily living. Investigations revealed massive proteinuria suggestive of nephrotic syndrome, high resolution computed tomography suggestive of interstitial lung disease and skin biopsy revealed epidermolysis bullosa. The patient was given supportive treatment and her peripheral blood sample was subjected to next generation sequencing, which revealed a homozygous 3’ splice site mutation in intron 13 of the ITGA3 gene. Our sequencing efforts suggested the presence of an ITGA3 psuedogene. This pseudogene could express a truncated but partially functional version of the ITGA3 protein, which would explain the milder clinical symptoms in this patient. We are investigating this possibility. In summary, we describe a rare variant of the ILNEB syndrome associated with a homozygous mutation in the ITGA3 gene. The presentation of milder phenotypes in this case is distinct from previously reported instances of this syndrome where all patients died by 19 months of age from multi-organ failure.
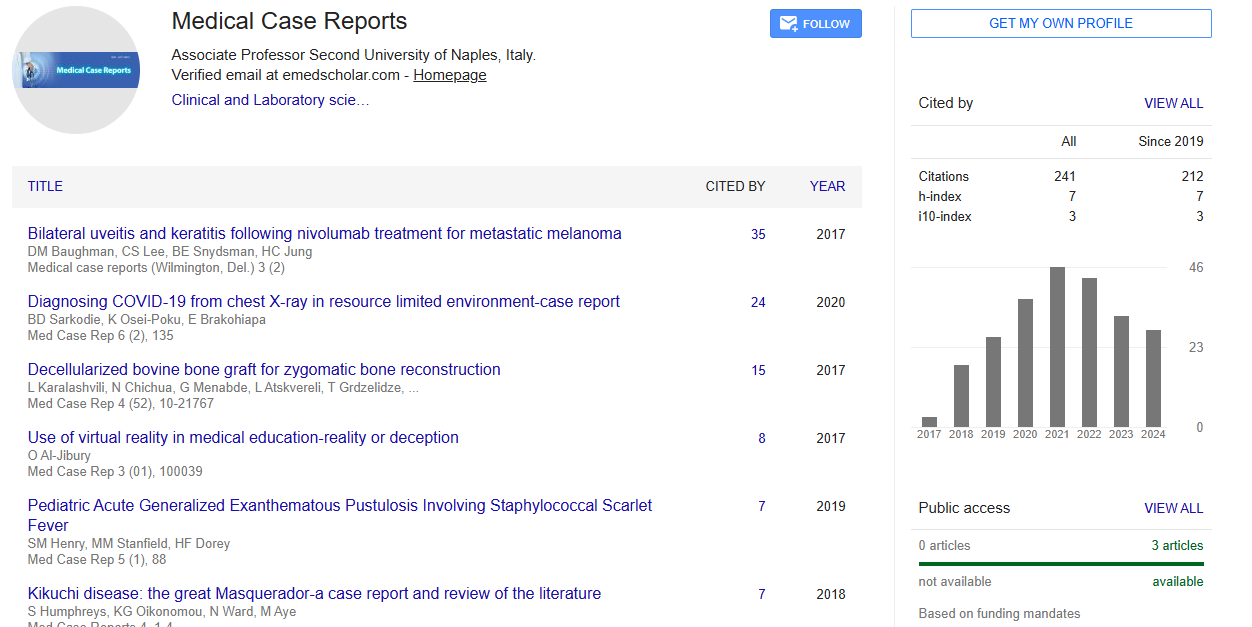
Google Scholar citation report
Citations : 241
Medical Case Reports received 241 citations as per Google Scholar report
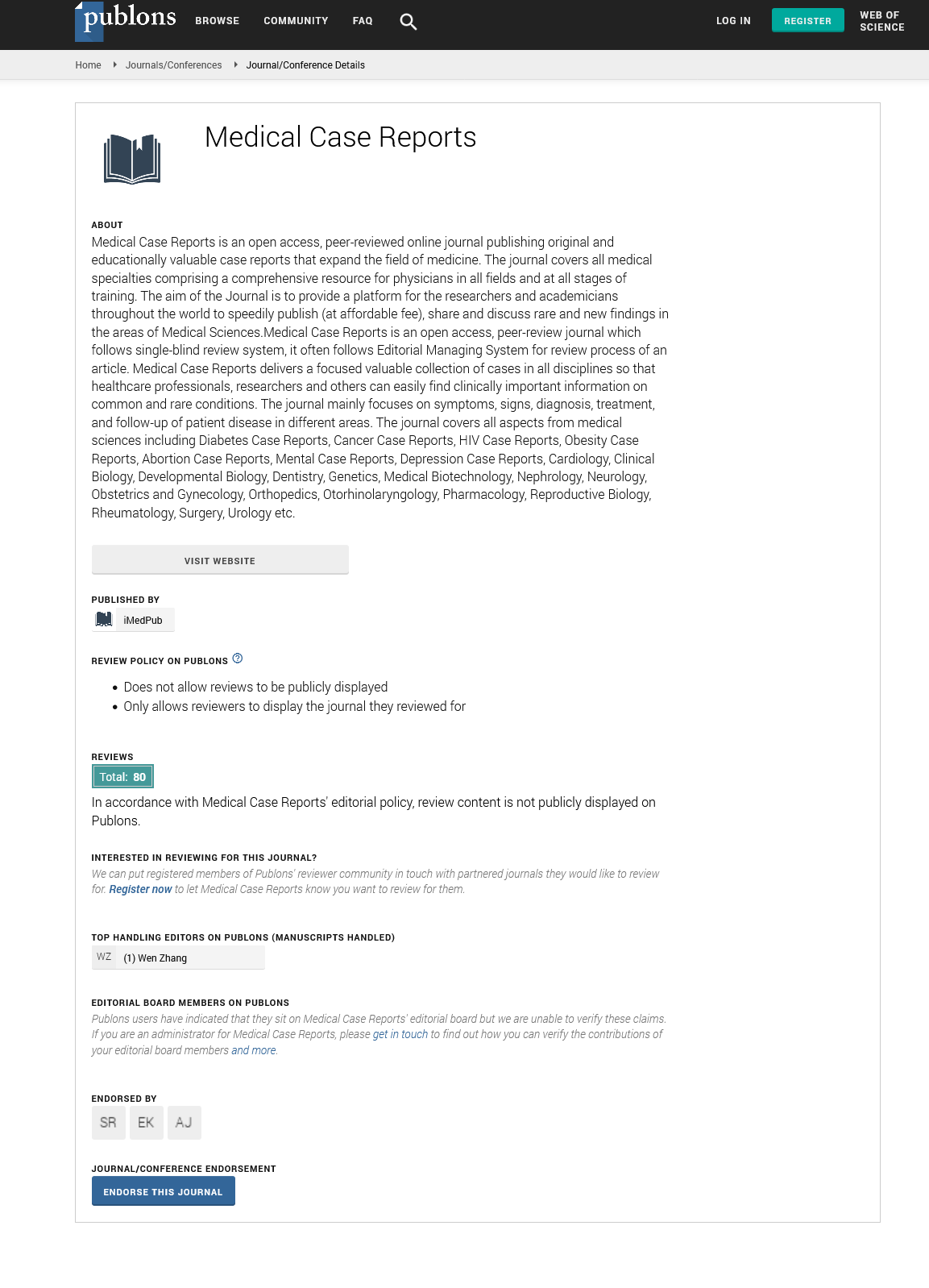
Medical Case Reports peer review process verified at publons
Abstracted/Indexed in
- Google Scholar
- China National Knowledge Infrastructure (CNKI)
- Cosmos IF
- Directory of Research Journal Indexing (DRJI)
- WorldCat
- Publons
- Secret Search Engine Labs
- Euro Pub
Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences