Ionic Diffusion and Proton Transfer in Aqueous Solutions under an Electric Field: State-of-The-Art
Fabrizio Creazzo*
University of Paris-Saclay, France
- *Corresponding Author:
- Fabrizio Creazzo
Researcher
University of Paris-Saclay, France
Tel: +33 637250092
E-mail: fabrizio.creazzo@univ-evry.fr
Received Date: August 22, 2017; Accepted Date: August 23, 2017; Published Date: August 31, 2017
Citation: Creazzo F (2017) Ionic Diffusion and Proton Transfer in Aqueous Solutions under an Electric Field: State-of-The-Art. J Mol Sci. 1:2.
Most of the properties and anomalies describing the behavior of water are somehow related to the hydrogen bonded (H-bonded) network [1-3]. Albeit the features of H-bonds have been investigated and depicted by an impressive amount of research, the way in which some external conditions–such as the inclusion of ionic species–affect the three-dimensional H-bonds arrangement is wrapped up in a high degree of uncertainty.
If, on one hand, the presence of solvated ions cannot be avoided even in ultra-pure water samples, on the other hand, the lack of scientific consensus about the ion-induced microscopic effects on the water structure is representative of the practical challenges faced when investigating electrolyte solutions [4,5]. However, the indisputable role played by a few atomic charged species both in biology (i.e., Na+, Cl–, Mg2+, Ca2+, etc.) [6-8] and in industry (e.g., Li+ batteries) [9] requires impelling and massive scientific efforts. In fact, besides the well-known Hofmeister series [10], hydrated ionic species finely rule the selectivity of cell membranes [6,7], being thus responsible of complex processes such as the nerve pulse generation. On the other hand, aqueous solutions represent the prototype of electrolytic batteries.
In all cases, a subtle balance between electrostatics, quantum mechanics (i.e., partial orbital sharing), and thermodynamics governs the delicate behaviour of the hydration process. The complexity of the problem is witnessed, inter alia, by the fact that there is no general consensus on the spatial extent of the effects induced by the inclusion of an ion in bulk water [11-13].
Recent ab initio calculations [14] have shown that the presence of a chaotrope species such as Cl− does not have any effect on the orientation of water dipoles beyond the first hydration shell, whereas detectable perturbations–perhaps extremely small and unable to affect biological phenomena–have been observed in the polarizability of the water molecules at longer distances.
Additionally, the lack of a wide consensus on the typical coordination numbers characterizing the ionic first solvation shell is thoroughly recorded in the literature [4]. From an experimental perspective, the identification of this quantity is a very hard task for small ions such as Li+ and, recently, new ionic radii for this species and for Na+ have been proposed [4] by joining the advantages stemming from Large Angle X-ray Scattering (LAXS) and double Difference Infrared Spectroscopy (DDIR). In this respect, ab initio Molecular Dynamics (AIMD) [8,14] and QM/MM [15] computational techniques have proven their reliability in reproducing the ion-induced structural changes in aqueous solutions, thus becoming an invaluable tool for the characterization of electrolyte solutions at a molecular level.
In particular, it seems that at low-to-moderate concentrations the ions may replace water molecules in the aqueous H-bonded structure, by following the same ‘‘water rules’’. This example proves that classical molecular dynamics may fail in dealing with delicate local electrostatic balances and that first-principles approaches are necessary not only for a correct microscopic characterization of these phenomena but also in order to improve the models on which classical force fields rely.
Indeed, although sixty years ago concepts such as kosmotrope and chaotrope have been introduced to characterize the perturbation produced by a given ion on the H-bond network of water [16,17], and notwithstanding the fact that these notions were supported by classical molecular dynamics simulations [18-20], they have recently been blunted by an AIMD study [21]. Ionic conductivities are determined by applying an oriented external static electric field to electrolyte solutions. When an external electric field is applied, the situation is even tougher.
In the low field strength regime and within the Kohlrausch’s law of independent migration of ions (i.e., in the limit of infinite dilution), the mobilities of the alkali metal cations are well-established and can be easily related to their respective ionic sizes [22] i.e. the bigger the cation the larger the mobility. However, at finite molarities and for stronger field intensity regimes the overall situation may dramatically change. Field intensities of the order of 1 V/Å and even stronger were detected at the atomic sites of the water molecules hydrating Na+ and Cl– ions [23], suggesting that for moderate-to-intense field strengths more complicated phenomena may be relevant in describing the ionic diffusion. Moreover, field intensities of about 0.30 V/Å are able to induce the molecular dissociation of water and proton transfers along the H-bonded network [24-27] via the well- known proteolysis reaction:
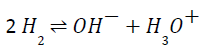
This latter process plays a crucial role in many disparate domains, from neurobiology to electrolytic batteries and hydrogen-based technology [28,29]. Thus, it can be expected that a subtle interplay between the two deeply different mechanisms of protonic migration, on one hand, and of standard ionic diffusion, on the other, rules the complex dynamics of electrolytic solutions subjected to intense field strengths.
References
- Debenedetti PG (2006) Metastable liquids-concepts and principles. Princeton University Press, Princeton, NJ, USA.
- Franks F (2000) Water: a matrix of life (2nd edn.). Royal Society of Chemistry, Cambridge.
- Brovchenko I, Oleinikova A (2008) Multiple phases of liquid water. ChemPhysChem 9: 2660-2675.
- Mahler J, Persson I (2012) A study of the hydration of the alkali metal ions in aqueous solution. Inorg Chem 51: 425-438.
- Smirnov PR, Trostin VN (2006) structure of the nearest surrounding of the Li+ ion in aqueous solution of its salts. Russ. J Gen Chem 76: 175-182.
- Hille B (2001) ion channels of excitable membranes (3rd edn.). Sinauer Associates, Inc. Publishers, Sunderland, MA, USA.
- Zhou Y, MacKinnon R (2003) the occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Bio 333: 965-975.
- Bankura A, Carnevale V, Klein ML (2013) Hydration structure of salt solution from ab initio molecular dynamics. J Chem Phys 138: 014501.
- Tarascon JM, Armand M (2001) issues and challenges facing rechargeable lithium batteries. Nature 414: 359-367.
- Kunz W, Henle J, Ninham BW (2004) “Zur Lehre von der Wirkung der Salze” (About the Science of the Effects if Salts): Franz Hofmeister’s historical papers. Curr Opin Colloid Interface Sci 9: 19-37.
- Stryer L, Haugland RP (1967) Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci USA 68: 719-726.
- Enderby JE (1995) Ion solvation via neutron-scattering. Chem Soc Rev 24: 159-168.
- Collins KD, Neilson GW, Enderby JE (2007) Ions in water: characterizing the forces that control chemical processes and biological structure. Biophys Chem 128: 95-104.
- Gaiduk AP, Galli G (2017) Local and global effects of dissolved sodium chloride on the structure of water. J Phys Chem Lett 8: 1496-1502.
- Rowley CN, Roux B (2012) The solvation structure of Na+ and K+ in liquid water determined from high level ab initio molecular dynamics simulations. J Chem Theo Comp 8: 3526-3535.
- Gurney RW (1953) Ionic Processes in Solution. McGraw-Hill: NY, USA.
- Frank HS, Wen WY (1957) Structural aspects of ion-solvent interaction in aqueous solutions: a suggested picture of water structure. Discuss Farad Soc 24: 133-140.
- Galamba N (2013) On the effects of temperature, pressure, and dissolved salts on the hydrogen- bond network of water. J Phys Chem B 117: 589-601.
- Reagan MT, Harris JG, Tester JW (1999) Molecular simulations of dense hydrothermal nacl-h2o solutions deom subcritical to supercritical conditions. J Phys Chem B 103: 7935-7941.
- Renou R, Ding M, Zhu H, Szymcyk A, Malfreyt P, et al. (2014) concentration dependence of the dielectric permittivity, structure, and dynamics of aqueous NaCl solutions: comparison between the drude oscillator and electronic continuum models. J Phys Chem B 118: 3931-3940.
- Ding Y, Hassanali A, Parrinello M (2014) Anomalous water diffusion in salt solutions. Proc Natl Acad Sci USA 111: 3310-3315.
- Wright MR (2007) An Introduction to aqueous electrolyte solutions. Wiley: Chichester, England.
- Sellner B, Valiev M, Kathman SM (2013) Charge and electric field fluctuations in aqueous NaCl electrolytes. J Phys Chem B 117: 10869-10882.
- Stuve EM (2012) Ionization of water in interfacial electric fields: an electrochemical view. Chem Phys Lett pp: 519-520, 1-17.
- Saitta AM, Saija F, Giaquinta PV (2012) Ab Initio molecular dynamics study of dissociation of water under an electric field. Phys Rev Lett.
- Cassone G, Creazzo F, Giaquinta PV, Saija F, Saitta AM (2016) Ab initio molecular dynamics study of an aqueous NaCl solution under an electric field. Phys Chem Chem Phys 18: 3164-23173.
- Hammadi Z, Descoins M, Salanon E, Morin R (2013) Proton and light ion nanobeams from field ionization of water. J Appl Lett 101: 110-243.
- Kaila K, Ransom BR (1998) pH and brain function; In: Kaila K, Ransom BR (eds.) Wiley, NY, USA.
- Zoulias EI (2008) Lymberoupolos, N. hydrogen based autonomous power systems. Springer, London.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences