ISSN : 2634-7814
Journal of Chemical Biology & Pharmaceutical Chemistry
Development of Novel Isoxazole Bearing Lead against Alzheimer's disease By Docking Guided Virtual Screening and De Novo Ligand Design
Souvik Basak*1, Puja Mishraa1 and Arup Mukherjee
1Department of Allied Health Sciences, Dr. B.C. Roy College of Pharmacy & Allied Health Sciences, Durgapur, WB, India
2Department of Biotechnology, Maulana Abul Kalam Azad University of Technology, WB, India
*Corresponding author: Souvik Basak, Department of Allied Health Sciences, Dr. B.C. Roy College of Pharmacy & Allied Health Sciences, Durgapur, WB, India; Tel No: +91-9051226973; E-mail: souvik_basak1@yahoo.com
Received date: August 04, 2020; Accepted date: August 13, 2021; Published date: August 23, 2021
Citation: Souvik B (2020) Development of Novel Isoxazole Bearing Lead against Alzheimer’s Disease By Docking Guided Virtual Screening and De Novo Ligand Design. Pharm. Chem. J Vol: 4 No: 4.
Abstract
Abnormal amyloid β protein (Aβ) aggregation is the initiating process of Alzheimer’s disease. Anti-amyloid compounds must inhibit such accumulation by stabilizing soluble Aβ oligomers by forming hydrogen bonds and π-π stacking. Recent reports suggest that curcumin inhibits Aβ fibril by producing structural changes in the salt bridge of Aβ protein and modifying fibril conformation. Curcumin analogs with pyrazole, isothiazole and isooxazoles exhibit considerable binding affinity comparable to that of curcumin. Multiple group replacement by various isosteres in curcumin gave significant results. In silico docking studies of compound (28) with both Aβ protein and β-secretase enzyme (BACE1) was found to be higher than that of standard. De novo ligand design generated best suitable derivative of the selected lead by iterations. Both compound (28) and de novo designed lead offspring reached the binding pocket of both the proteins, passed Lipinski’s rule of five and in silico toxicity testing by admetSAR.
Keywords: Aβ fibril, curcumin, isooxazole, docking, de novo ligand design
Introduction
Alzheimer’s disease (AD) is defined as a progressive neurodegenerative disorder, characterized by gradual loss of cholinergic neurons and accumulation of Amyloid-β (Aβ) protein in the brain areas like cortex and hippocampus [1]. The disease onset starts with short term memory impairment that gradually progresses to complete loss of cognitive function and ultimately death. Individuals who suffer from Alzheimer have numerous Senile Plaques (SPs) and Neuro Fibrillary Tangles (NFTs) characterized by neural cell loss and vascular damage. Aβ that build the connection between the nerve cells are the cause of the above condition.
Abnormal processing of Amyloid Precursor Protein (APP), a trans-membrane protein present on the surface of neurons generates Aβ which is the main cause of Alzheimer’s disease [2]. APP is cleaved by a series of enzymes called secretase. In normal physiology, α-secretase and γ-secretase cleaves APP to give p3 domain in the extracellular matrix and APP intracellular domain (AICD) in the cytoplasm. In case of Alzheimer’s disease instead of α-secretase, an abnormal β-secretase cleaves APP followed by γ-secretase and produces Aβ in the extracellular domain along with AICD in the cytoplasm [3]. Aβ mostly contains 40 (Aβ40) or 42 (Aβ42) amino acid residues of which Aβ42 is susceptible to aggregation[4].By unknown mechanism, monomeric Aβ polymerizes, forms long insoluble fibrillary aggregates, and accumulates as extra cellular deposits called as senile plaques. Increased levels of metals like Zn, Cu, Fe are also linked to the development of Alzheimer’s disease [5]. Consequently, inhibiting abnormal Aβ aggregation is considered to be a potential therapeutic target for AD.
Aβ42 monomers join to form oligomers by nucleation dependent polymerization. Oligomeric structure of Aβ attains stability by increasing the number of hydrogen bonds, peptide interactions and hydrophobic contacts [6]. Aβ42 is a short peptide sequence with N-terminal β1 (Val 18-Ser 26) and C-terminal strand β2 (Ala 31- Ala 42). The salt bridge (Asp 23-Lys 28) adds extra stability to the oligomers and a hydrophobic core (KLVFFAE) is required for dimerization with other monomers. [7].
Earlier studies have reported that polyphenols and their derivatives acts as anti-aggregating and destabilizing agent [8].Curcumin is a polyphenolic compound found to be neuro-protective in a transgenic animal model for Alzheimer’s disease[9]. Various in vitro tests shows that Curcumin inhibits β-secretase activity, Aβ induced inflammation and Aβ aggregation[10]. Curcumin binds to Aβ oligomers and inhibits formation both in vitro and in vivo[11].Curcumin binds to transition metals like Fe, Cu and reduces Aβ toxicity via metal chelation[12]. Curcumin in lesser dose reduces amyloid induced cell death and reactive oxidative stress [13].
In compliance with the earlier reports, it is known that a molecule forming hydrogen bonding interaction and π- π interaction could be a potential compound [14]. Thus, new chemical library of molecules with substitution in curcumin were designed aiming at enhanced stability and greater binding efficacy. Fragment based ligand design was achieved by isosteric replacement of various functional groups of curcumin. The chemical library was then docked sequentially to Aβ protein and screened based on their docking scores and drug likeliness property. Selected docked leads were redocked against β-secretase (BACE1) focussing on dual inhibition of fibrillation mechanism.
MATERIALS AND METHODS
Materials
Ligands were drawn using ChemDraw Pro 12.0 (Cambridge Soft, Cambridge Park Drive, Cambridge) and docking scores were obtained AutoDock Vina 4.2.6 (The Scripps Research Institute, LaJolla, California). The ligand-protein interaction of the output file obtained after docking was visualized in BIOVIA Discovery Studio Visualizer 2020 (Dassault Systèmes, San Diego, USA). Lead modification of the best fitting ligand was achieved using eLEA3D (https://chemoinfo.ipmc.cnrs.fr/LEA3D/index.html). Drug likeliness of the compounds showing comparative docking scores was checked via Molinspiration (https://www.molinspiration.com/cgi-bin/properties) and in silico toxicity was done in admetSAR 2.0 (http://lmmd.ecust.edu.cn/admetsar1/predict/).
Development of chemical library
All out – In our study, Curcumin ((1E, 6E)-1, 7-bis (4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione) is taken as the lead compound and stable analogs of curcumin are designed against fibrillation by substitutions in (A) and (B) as shown (Fig. 1).
Structural activity relationship of curcumin shows phenolic hydroxy group is essential for inhibiting Aβ aggregation. The curcumin analogs should have two aromatic phenolic groups with seven carbon linker stacked in between is essential for its optimum activity. Monosubstitutions in methoxy curcumin analogues showed greater inhibitory activity as compared to disubstituted ones [15] [16][17].
Thus chemical library was prepared fixing the phenolic ends and seven carbon linkers but modifying the 1, 3-dicarbonyl moiety (A) and methoxy group (B) was carried out. 1,3-dicarbonyl moiety is su In our study, Curcumin ((1E, 6E)-1, 7-bis (4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione) is taken as the lead compound and stable analogs of curcumin are designed against fibrillation by substitutions in (A) and (B) as shown (Fig. 1).
Structural activity relationship of curcumin shows phenolic hydroxy group is essential for inhibiting Aβ aggregation. The curcumin analogs should have two aromatic phenolic groups with seven carbon linker stacked in between is essential for its optimum activity. Monosubstitutions in methoxy curcumin analogues showed greater inhibitory activity as compared to disubstituted ones [15] [16][17].
Thus chemical library was prepared fixing the phenolic ends and seven carbon linkers but modifying the 1, 3-dicarbonyl moiety (A) and methoxy group (B) was carried out. 1,3-dicarbonyl moiety is substituted by heterocyclic isosteres isooxazoles, pyrazole and isothiazole as they are known to restrict rotational freedom and improve pharmacological properties of curcumin[18][12][19].
Accordingly ligands were designed based on three regimes to provide molecules with unique properties as compared to curcumin. The three regimes are firstly substitution in (A) by pyrazole and its derivatives (compound 1-13), secondly substitution in both (A) and (B) by pyrazole and its derivatives and hydroxy group respectively (compound 14-26), and thirdly substitution in both (A) and (B) by isothiazole and isooxazoles and methoxy isosteres respectively (compound 27-39).bstituted by heterocyclic isosteres isooxazoles, pyrazole and isothiazole as they are known to restrict rotational freedom and improve pharmacological properties of curcumin[18][12][19].
Accordingly ligands were designed based on three regimes to provide molecules with unique properties as compared to curcumin. The three regimes are firstly substitution in (A) by pyrazole a In our study, Curcumin ((1E, 6E)-1, 7-bis (4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione) is taken as the lead compound and stable analogs of curcumin are designed against fibrillation by substitutions in (A) and (B) as shown (Fig. 1).
Structural activity relationship of curcumin shows phenolic hydroxy group is essential for inhibiting Aβ aggregation. The curcumin analogs should have two aromatic phenolic groups with seven carbon linker stacked in between is essential for its optimum activity. Monosubstitutions in methoxy curcumin analogues showed greater inhibitory activity as compared to disubstituted ones [15] [16][17].
Thus chemical library was prepared fixing the phenolic ends and seven carbon linkers but modifying the 1, 3-dicarbonyl moiety (A) and methoxy group (B) was carried out. 1,3-dicarbonyl moiety is substituted by heterocyclic isosteres isooxazoles, pyrazole and isothiazole as they are known to restrict rotational freedom and improve pharmacological properties of curcumin[18][12][19].
Accordingly ligands were designed based on three regimes to provide molecules with unique properties as compared to curcumin. The three regimes are firstly substitution in (A) by pyrazole and its derivatives (compound 1-13), secondly substitution in both (A) and (B) by pyrazole and its derivatives and hydroxy group respectively (compound 14-26), and thirdly substitution in both (A) and (B) by isothiazole and isooxazoles and methoxy isosteres respectively (compound 27-39).nd its derivatives (compound 1-13), secondly substitution in both (A) and (B) by pyrazole and its derivatives and hydroxy group respectively (compound 14-26), and thirdly substitution in both (A) and (B) by isothiazole and isooxazoles and methoxy isosteres respectively (compound 27-39).

Figure1. Chemical modifications in Curcumin (keto form)
Molecular Docking studies and selection of the lead
After ligand preparation, 42 residue β-pleated Aβ protein was selected and downloaded from Protein Data Bank (www.rcsb.org, PDB ID: 2BEG)[20]. The crystallographic structure of protein was processed by adding Kollman charges, merging non polar hydrogen and removing water molecules. A grid box enclosing the protein was made, having specific dimension (center_x=0.385; center_y=0.250; center_z= -8.949). Both ligand and protein was converted to PDBQT format in AutoDock Tools 4. The designed chemical library was screened by docking the ligand and Aβ protein using AutoDock Vina 4.2.6 (The Scripps Research Institute, La Jolla, California)[21]. Apparently the leads showing best binding efficiency with 2BEG and passing drug likeliness property were selected and redocked with BACE1 enzyme protein(beta site amyloid precursor protein cleaving enzyme) (www.rcsb.org, PDB ID: 2ZHT)[22][23]. BACE1 are transmembrane aspartic protease that leads to accumulation of Aβ, thus acts as a potential therapeutic target for AD[24]. The co-crystallized ligand with 2ZHT was deleted and the water molecules were deleted and a grid box having specific dimension (center_x=64.898; center_y=46.972; center_z= -0.369) was made that enclosed the entire protein. The compound giving apparent results for both the protein 2BEG and 2ZHT are selected as the best lead that can act as dual inhibitors of both Aβ and β-secretase.
De Novo Ligand Design
De novo ligand design provided lead modification of the best lead obtained after docking against 2BEG and 2ZHT. Such ligand design is carried out using eLEA3D which generates offsprings that suitably binds to the protein. It requires .pdb file of the protein and it’s binding site information as input. The protonated protein file (protein.mol2) having only amino acid is used to perform docking in PLANTS algorithm. A scaffold structure (ligand.sdf) along with its 3D co-ordinates is uploaded to optimize the best lead. The atom number that is to be linked was mentioned to provide selective binding of the ligand. De novo ligand design is performed by cross over mutation between the generated offsprings and each offspring is generated by each round of design parameters. The best derivative of the lead was selected based on their binding affinity and fitting score on the template lead.
Evaluation of Lipinski’s Rule of Five
Permission After docking, drug likeliness of selected lead was evaluated by Lipinski’s rule of five. The SMILES format of the selected lead was used as input in MOLINSPIRATION server and the Lipinski’s rule of five (log P ≤ 5, molecular weight ≤ 500 Da, nON ≤ 10, nOHNH ≤ 5) was estimated. If the lead passes the mentioned parameters then the lead is likely to be orally active drug in humans.
Evaluation of Admetsar of The Selected Lead
Biavailable drugs must be screened for pharmacokinetic parameters such as ADMET (absorption, distribution, metabolism, excretion and toxicity). ADMET prediction helps to develop superior quality lead. The SMILES of the selected lead was given as input in admetSAR server and in silico toxicity data was produced to estimate their property to become a drug [25].
Results and discussions
Molecular docking studies of compound library and curcumin with 2BEG and 2ZHT
Literature survey of Curcumin, elucidated the importance of terminal phenol group, seven carbon linker, poly hydroxy groups and 1, 3-dicarbonyl moiety in its structure[18][12]. Enhanced stability was ensured by replacing (A) the 1, 3-dicarbonyl moiety by pyrazole and its derivatives, isooxazole and isothiazole. Introducing bioisosteres of –OCH3 (B) such as –OH, –F, -CF3, -CF2CH3 along with heterocyclic moiety was aimed to improve binding interaction with receptor. Docking scores of such modifications mostly ranged in between -5.9 to -7.1 Kcal/mol(Table 1) which is even more than that of standard curcumin (-5.5 Kcal/mol).
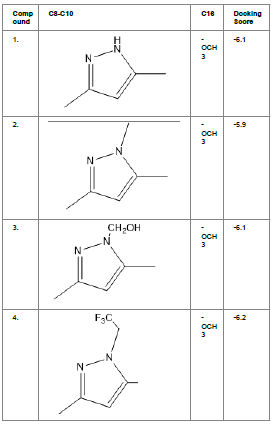
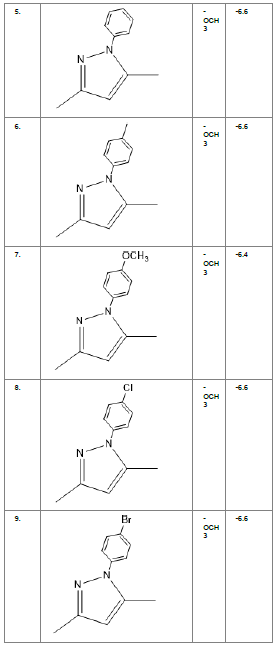
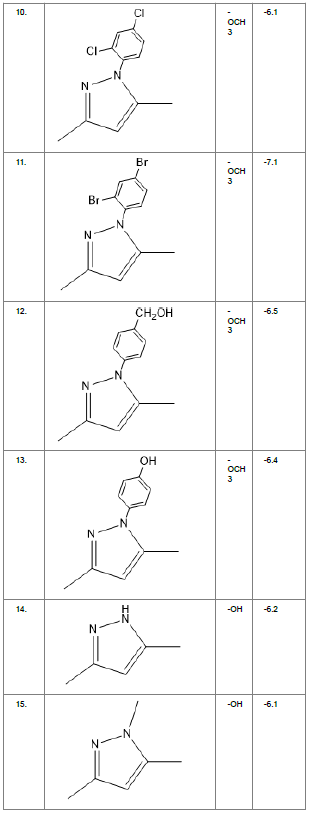
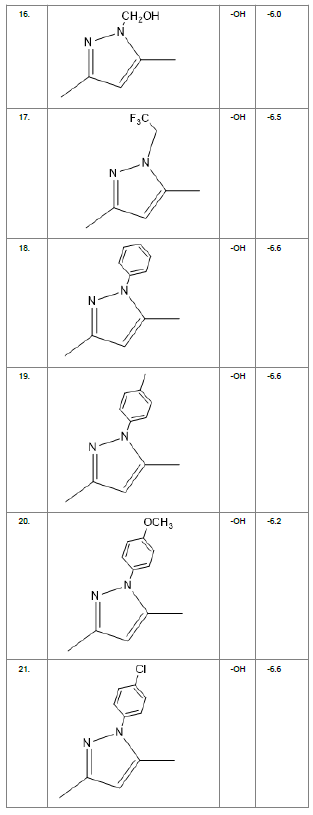
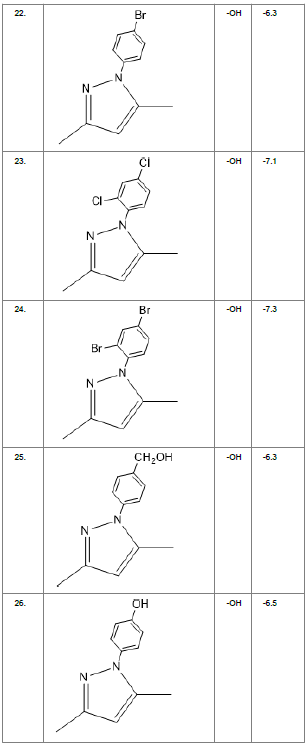
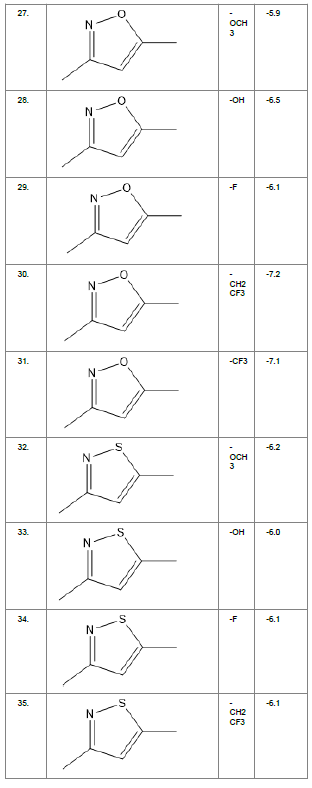
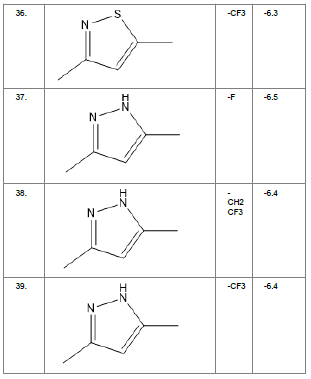
Table1. Chemical library showing the modifications along with their docking scores
Compound 24 gave the highest docking score of -7.3 Kcal/mol and Compound 30 show the docking score of -7.2 Kcal/mol. Docking score of compound 31, 11, 23 exhibits binding efficiency of -7.1 Kcal/mol. A large number of compound 5, 6, 8, 9, 18, 19 and 21, from the chemical library so designed gave a docking score of -6.6 Kcal/mol. Binding efficiency of -6.5 Kcal/mol was observed for compound 12,17, 26, 28 and 37. The docking results of 17 compounds gave acceptable results after docking against the protein 2BEG.
Evaluation of drug likeliness of all the 17 screened compounds was observed in MOLINSPIRATION server. Among the above screened compounds only 4 compounds confirmed drug like properties after passing the Lipinski’s rule of five. Compound 12, 26, 28, 17 qualified the bioavailability related factors as shown (Table 2).
| Compound | Docking scores | miLogP | MW (Da) | nON | nOHNH |
|---|---|---|---|---|---|
| -24 | -7.3 | 7.03 | 584.26 | 6 | 3 |
| -30 | -7.2 | 5.9 | 399.39 | 5 | 2 |
| -31 | -7.1 | 5.81 | 403.36 | 5 | 2 |
| -11 | -7.1 | 7.33 | 598 | 6 | 2 |
| -23 | -7.1 | 6.76 | 495 | 6 | 3 |
| -5 | -6.6 | 5.57 | 440.5 | 6 | 2 |
| -6 | -6.6 | 6.02 | 454.53 | 6 | 2 |
| -8 | -6.6 | 6.25 | 474.94 | 6 | 2 |
| -9 | -6.6 | 6.38 | 519.39 | 6 | 2 |
| -18 | -6.6 | 5.27 | 426.47 | 6 | 3 |
| -19 | -6.6 | 5.72 | 440.5 | 6 | 3 |
| -21 | -6.6 | 5.95 | 460.92 | 6 | 3 |
| -12 | -6.5 | 4.91 | 470.52 | 7 | 3 |
| -26 | -6.5 | 4.79 | 442.47 | 7 | 4 |
| -28 | -6.5 | 4 | 351.36 | 6 | 3 |
| -17 | -6.5 | 4.92 | 432.4 | 6 | 3 |
Table 2. Drug likeliness list for the screened compounds
*miLogP-Molinspiration LogP, MW-Molecular Weight, nON-number of hydrogen bond acceptors, nOHNH-number of hydrogen bond donors
Compound 12, 26, 28, 17 and the standard (curcumin) were redocked over the BACE1 peptide (2ZHT). Compound 12 and 26 showed binding efficiency of -6.8 Kcal/mol and -7.3 Kcal/mol respectively which is less as compared to standard (-7.9 Kcal/mol). Rest two compounds 28 and 17 give better docking scores of -8.8Kcal/mol and -8.7 Kcal/mol respectively. Compound (28) (isooxazole isostere replacement in A and –OH in B) is the best selected lead acting as dual inhibitor (Fig. 2).

Figure2. Compound (28) (Best selected lead from 2BEG, 2ZHT & drug likeliness)
Standard curcumin and compound (28) shows similar interactions with protein 2BEG. Standard Curcumin (Fig. 3A) and Compound (28) shows vander waals interaction with Gly A: 38, conventional hydrogen bond with Val A:39, π-π stacking with Phe A:19 and π-sigma with Val A:40 (Fig. 3B). Compound (28) interacts with KLVFFAE fragment (Lys 16-Glu 22) which is essential for dimerization of the monomers by π-π stacking with Phe A:19. Amino acid residues from 38-42 show hydrogen bonding interactions with 2BEG and can be a potential lead.

Figure3.Comparison of 2BEG binding site A. Standard curcumin B. Compound (28)
Standard Curcumin (Fig. 4A) and compound (28) exhibits greater interaction with the active site (Fig. 4B).Standard curcumin forms conventional hydrogen bond with Arg A: 307 and carbon hydrogen bond Glu A: 339. Similarly, conventional hydrogen bond with Arg A: 157 and carbon hydrogen bond Glu A: 339. Both standard and (28) are involved in π-π stacking with Trp A: 227. Consequently, Isooxazole replacement with –OH group (28) gives best result that shows that the cyclic ring in the symmetric region gives more stability to the structure of curcumin along with similar hydrogen bonding interactions and binding efficiency.

Figure4. Comparison of 2ZHT binding sites A. Standard curcumin B. Compound (28)
De novo ligand design
The de novo ligand design picks a template fragment arbitrarily and places it in the binding site by placing different fragments into the centre of binding pocket. Such fragments are then joined together to form completely different offsprings of the best selected lead. Compound (28) derivatives were generated by de novo ligand design that ensures lead modification. This compound generates 10 offsprings of which offspring 6 (Compound (28_6), Fig. 5) shows the best fitting score of 56.83%. Derivatives from offspring (5, 8, 10, 9) show proportional results of 56.22%, 56.21%, 56.20%, 55.59% respectively. The Compound (28_6) so generated is redocked with both Aβ protein and BACE1 to observe it’s binding efficiency. Compound (28_6) gave a docking score of -6.6 Kcal/mol with 2BEG which is greater than the binding efficiency of compound (28). The conformer so generated was docked with BACE1 and showed comparable results of -7.3 Kcal/mol. Equivalent results of both Compound (28) and Compound (28_6) shows that the ligand so designed is chemically distinct.

Figure5. Chemical structure of Compound 28_6, ligand by de novo design
In Silico Toxicity Study Evaluation By Admetsar
Twelve Dual inhibitor (28) shall be proved to be an effective lead for further research only if the compound shows no toxicity. Thus in silico toxicity prediction using admetSAR 2.0[25] is performed that ensures it to be safe as they show blood brain barrier absorption and no mutagenicity or carcinogenicity (Table 3). In silico toxicity study of both compound (28) and compound (28_6) was carried out and both revealed promising results. Absorption and metabolism drug profile for standard, compound (28) and compound (28_6) was comparable. These compounds were neither hepatotoxic nor carcinogenic. Thus, best screened lead (28) and de novo designed lead offspring (28_6) proves itself to be promising lead for Alzheimer’s disease treatment as they can cross blood brain barrier with no added toxicity or drug drug interaction. For further reference, compound (28) and compound (28_6) are referred as compound I and Compound II.
| Parameter | Curcumin | Compound (28) | Compound (28_6) |
|---|---|---|---|
| BBB | - | + | - |
| Human oral bioavailability | - | - | - |
| Hepatotoxicity | - | - | - |
| Carcinogenecity | - | - | - |
| Mutagenicity | - | - | - |
| CYP2C9 inhibition | - | - | - |
| CYP3A4 inhibition | + | + | + |
| Steroid receptor binding | + | + | + |
| p-glycoprotein inhibition | + | - | + |
Table3. In silico toxicity evaluation by admit SAR
+ means toxicity found, - means no toxicity foun
Conclusion
Drug against AD should target to prevent Aβ accumulation and increase the clearance of Aβ. Targeting Aβ and BACE1 protease shall inhibit Aβ fibrillation by stabilizing the soluble Aβ conformer. Curcumin is selected as standard for drug design as they show various anti-fibrillating properties. Curcumin derivatives were designed to provide extra stability and docked against Aβ and BACE1 protease to develop dual targeting drug. Amidst the library so prepared, compound I (4-((E)-2-(3-((E)-4-hydroxy-3-methoxystyryl) isoxazol-5yl)-vinyl) benzene-1,2-diol) show reassuring results with higher docking scores of -6.5 Kcal/mol against Aβ protein (2BEG) and -8.8 Kcal/mol against BACE1 protease (2ZHT). It is noticed that compound I reaches the active site of 2BEG by showing hydrogen bond interactions and π-π stacking in the salt bridge region of the protein. The salt bridge (Asp 23-Lys 28) is essential for fibril destabilization. The same compound reaches the active site of BACE1 protease (Glu 339) and exhibits hydrogen bonding interactions thus showing that the molecule reaches the flap of the peptide. Pharmacophore overlap over compound I generated a compound II with best fitting score. Compound II was redocked with Aβ protein and 2BEG that gave docking scores of -6.6 Kcal/mol and -7.3 Kcal/mol respectively. De novo designed lead offspring (compound II) shows drug like properties with no relative toxicity. Apparently both compounds can act as dual inhibitor with no in silico toxicity and good bioavailability properties, which manifests the molecule to be a future lead for drug synthesis and research against AD.
Acknowledgement
The Authors are thankful to Dr. B.C. Roy College of Pharmacy & Allied Health Sciences, Durgapur, WB, and India for providing all the support for execution of the work.
REFERENCES
- Broe GA, Grayson DA, Creasey HM, Waite LM, Casey BJ, Bennett HP, et al.(2000) Anti-inflammatory drugs protect against Alzheimer disease at low doses. Arch Neurol;57:1586â??91.
- Bandyopadhyay S, Huang X, Lahiri DK, Rogers JT.(2010) Novel drug targets based on metallobiology of Alzheimerâ??s disease. Expert Opin Ther Targets;14:1177â??97.
- Strooper B De. Amyloid-beta precursor protein processing in neurodegeneration 2004:582â??8. https://doi.org/10.1016/j.conb.2004.08.001.
- Zhang C, Browne A, Divito JR, Stevenson JA, Romano D. Amyloid- β Production Via Cleavage of Amyloid- β Protein Precursor is Modulated by Cell Density 2010;22:683â??94. [5]Shcherbatykh, Carpenter. Shcherbatykh, I., Carpenter, D.O. 2007 The role of metals in the etiology of Alzheimerâ??s disease. J Alzh Dis 2007;11:191â??205.
- Ramshini H, mohammad-zadeh M, Ebrahim-Habibi A. Inhibition of amyloid fibril formation and cytotoxicity by a chemical analog of Curcumin as a stable inhibitor. Int J Biol Macromol 2015;78:396â??404.
- Han X, He G. Toward a Rational Design to Regulate β-Amyloid Fibrillation for Alzheimerâ??s Disease Treatment. ACS Chem Neurosci 2018;9:198â??210.
- Chan S, Kantham S, Rao VM, Palanivelu MK, Pham HL, Shaw PN, et al. Metal chelation, radical scavenging and inhibition of Aβ42 fibrillation by food constituents in relation to Alzheimerâ??s disease. Food Chem 2016;199:14â??24.
- Liu Y, Dargusch R, Maher P, Schubert D. (2008)A broadly neuroprotective derivative of curcumin. J Neurochem;105:1336â??45.
- Lakey-Beitia J, González Y, Doens D, Stephens DE, SantamarÃa R, Murillo E, et al. Assessment of Novel Curcumin Derivatives as Potent Inhibitors of Inflammation and Amyloid-β Aggregation in Alzheimerâ??s Disease. J Alzheimerâ??s Dis 2017;60:S59â??68. [11]Narlawar R, Baumann K, Schubenel R, Schmidt B. (2007)Curcumin derivatives inhibit or modulate beta-amyloid precursor protein metabolism. Neurodegener Dis;4:88â??93.
- Ahmad B, Borana MS, Chaudhary AP.(2017) Understanding curcumin-induced modulation of protein aggregation. Int J Biol Macromol;100:89â??96.
- Lin CF, Yu KH, Jheng CP, Chung R, Lee CI.(2013) Curcumin reduces amyloid fibrillation of prion protein and decreases reactive oxidative stress. Pathogens;2:506â??19.
- Online VA, Rabiee A, Ebrahim-habibi A, Ghasemi A, Nemat-gorgani (2013)M. Food & Function amyloid fi brillation in insulin.
- Curcumin N-, Ahsan N, Mishra S, Jain MK, Surolia A, et al. (2015) Gupta S. modulate toxicity of Wild type.
- Endo H, Nikaido Y, Nakadate M, Ise S, Konno H. Bioorganic & Medicinal Chemistry Letters Structure activity relationship study of curcumin analogues toward the amyloid-beta aggregation inhibitor. Bioorg Med Chem Lett 2014;24:5621â??6. https://doi.org/10.1016/j.bmcl.2014.10.076.
- Cui L, Wang S, Zhang J, Wang M, Gao Y, Bai L, et al. Effect of curcumin derivatives on hen egg white lysozyme amyloid fibrillation and their interaction study by spectroscopic methods. Spectrochim Acta - Part A Mol Biomol Spectrosc 2019;223:117365. https://doi.org/10.1016/j.saa.2019.117365.
- Narlawar R, Pickhardt M, Leuchtenberger S, Baumann K, Krause S, Dyrks T, et al. Curcumin-Derived Pyrazoles and Isoxazolesâ?¯: Swiss Army Knives or Blunt Tools for Alzheimer â?? s Diseaseâ?¯? 2008:165â??72. https://doi.org/10.1002/cmdc.200700218.
- Nurfina AN, Reksohadiprodjo MS, Timmerman H, Jenie UA, Sugiyanto D, Van Der Goot H. Synthesis of some symmetrical curcumin derivatives and their antiinflammatory activity. Eur J Med Chem 1997;32:321â??8. https://doi.org/10.1016/S0223-5234(97)89084-8.
- Taylor P, Kumar A, Srivastava S, Tripathi S, Singh SK. Journal of Biomolecular Structure and Dynamics Molecular insight into amyloid oligomer destabilizing mechanism of flavonoid derivative 2- ( 4 â?² benzyloxyphenyl ) -3-hydroxy-chromen-4-one through docking and molecular dynamics simulations 2015. https://doi.org/10.1080/07391102.2015.1074943.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. Software News and Updates AutoDock4 and AutoDockTools4â?¯: Automated Docking with Selective Receptor Flexibility 2009. https://doi.org/10.1002/jcc.
- Konno H, Endo H, Ise S, Miyazaki K, Aoki H, Sanjoh A, et al. Synthesis and evaluation of curcumin derivatives toward an inhibitor of beta-site amyloid precursor protein cleaving enzyme 1. Bioorganic Med Chem Lett 2014;24:685â??90.
- Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N.(2008) Crystal Structure of an Active Form of BACE1, an Enzyme Responsible for Amyloid β Protein Production. Mol Cell Biol;28:3663â??71.
- Patel S, Vuillard L, Cleasby A, Murray CW, Yon J, Technology A. Apo and Inhibitor Complex Structures of BACE ( b -secretase ) 2004:407â??16..
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al.( 2012)admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences